Disentangling latent representations of single cell RNA-seq
- 4 minsThis post summarizes the motivations and results of my recent pre-print:
Disentangling latent representations of single cell RNA-seq experiments
Jacob C. Kimmel
bioRxiv 2020.03.04.972166; doi: https://doi.org/10.1101/2020.03.04.972166
Can we disentangle transcriptional space?
At the beginning of 2019, I became fascinated with the field of disentanglement learning. Disentanglement learning attempts to learn a latent representation of high-dimensional data where the dimensions of the latent space correspond to generative factors, or important parameters in the process that generated the data. Biology is ripe with potential applications for these methods, including in biological image analysis and genomics.
Single cell genomics has grown rapidly over the past decade, and experiments now regularly capture thousands of single cell profiles from complex tissues containing many cell types. One of the first steps in the analysis of these data sets is dimensionality reduction – trying to find a few latent dimensions that capture the underlying biological structure of gene expression space. Classical techniques like principal component analysis of non-negative matrix factorization can identify latent variables that explain variation in the data, but these latent variables are often difficult to interpret.
In an ideal latent space, we might want some of the underlying biological processes that influence cell state [e.g. cell cycle state] to be captured in a single latent variable. Disentanglement learning hopes to derive just such a representation from data, and I wanted to see if those methods could likewise learn interpretable latent space from single cell RNA-sequencing data.
I ran a few preliminary experiments as a side project, summarized in the pre-print above.
Representational trade-offs
Building on the variational autoencoder (VAE) methods of Lopez et. al., I augmented a standard single cell VAE model with a modified training objective inspired by $\beta$VAE and Burgess et. al..
These experiments yielded two key results:
(1) Disentanglement training improves alignment of latent variables and generative factors
I evaluated the alignment of VAE latent variables with ground-truth generative factors in simulated data, and I evaluated alignment with cell identity programs and experimental conditions in real experimental data. Based on these metrics, I found that latent variables more closely map to generative factors in both simulated and real data when disentanglement methods are applied. In the two figures below, conditions with $\beta = 1$ correspond to standard VAE training, while $\beta > 1$ corresponds to disentanglement training. Notice that the best correlations with generative factors or classification accuracies are obtained when disentanglement training is applied.
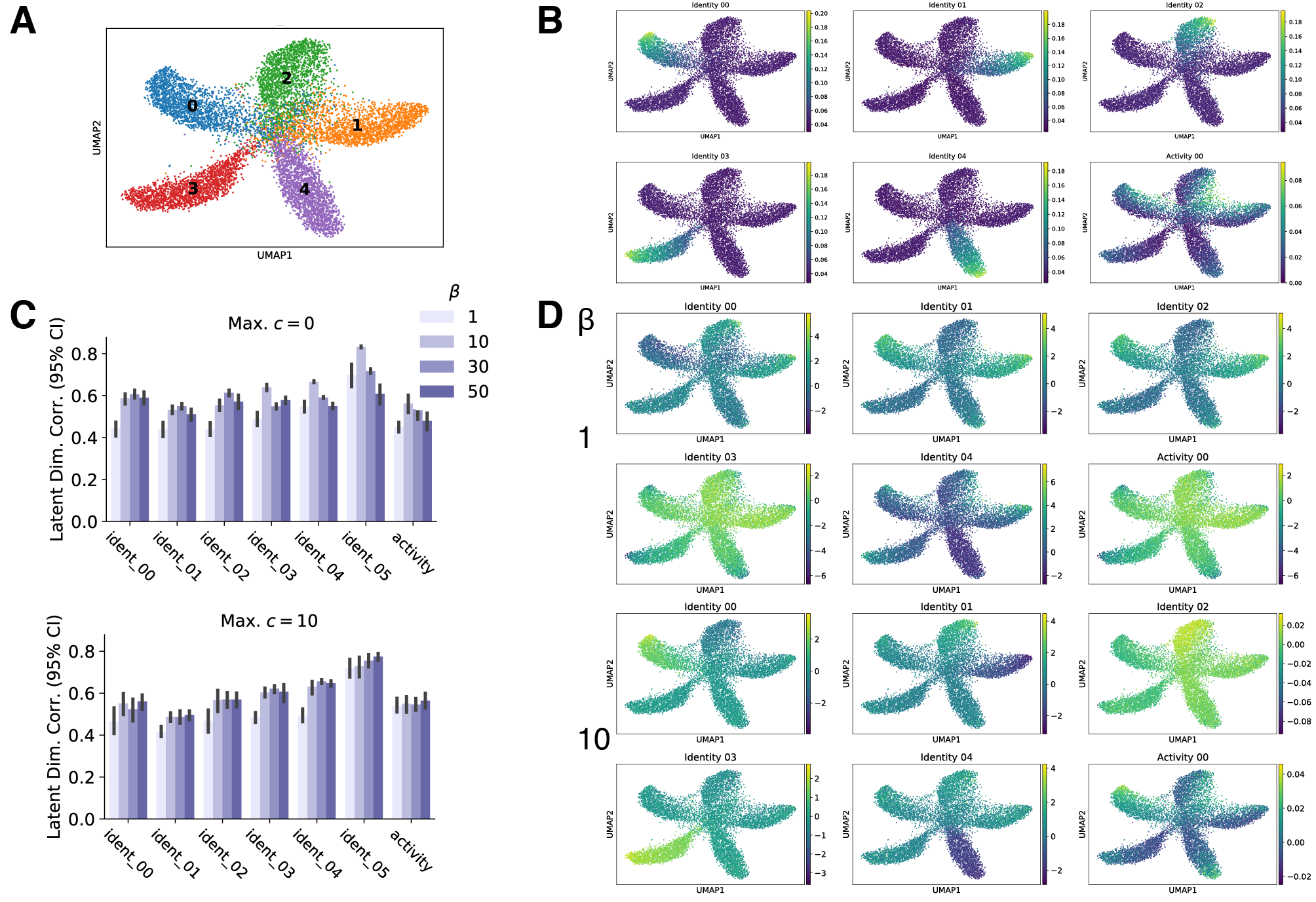
This results suggests that disentanglement techniques may be a promising method to learn interpretable representations of single cell transcriptomics data!
(2) Disentangled representations are less useful for some downstream tasks
Unfortunately, there’s a trade-off. The disentanglement training methods I used encourage the latent space to adhere closely to a prior distribution. In this case, our prior is a simple unit Gaussian $\mathcal{N}(0, \mathbf{I})$. This prior encourages the latent variables to be independent of one another, which is good for interpretability, but it also constrains the latent representation to a distribution that is highly unlikely to reflect real transcriptional space. Notice that cell type recovery by unsupervised clustering decreases when we apply disentanglement training in panel C below.
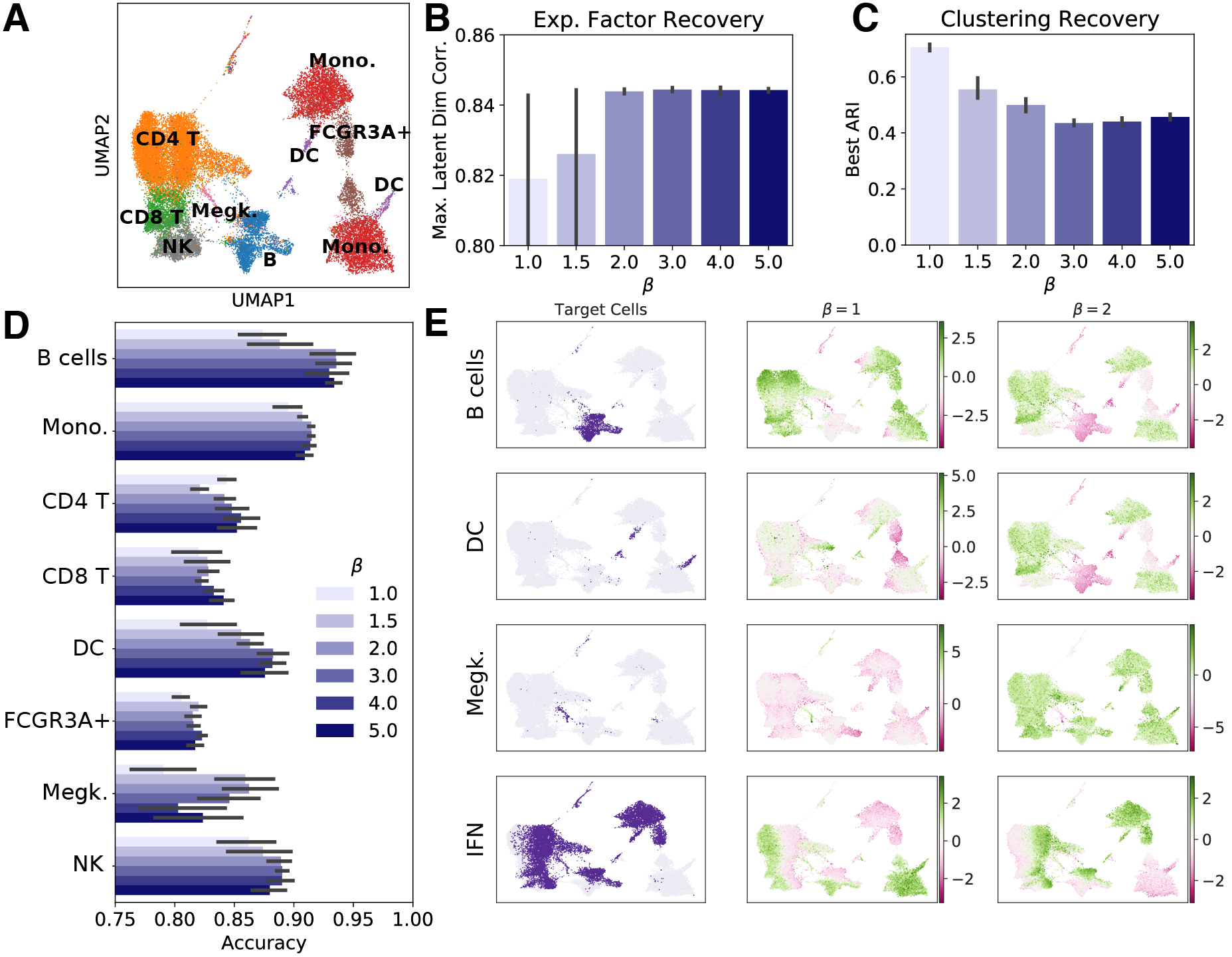
I saw this trade-off when performing downstream cell type clustering. In disentangled latent spaces, I saw worse clustering performance than in entangled representations. This suggests that while disentanglement training has benefits for interpretability, it may harm performance on some other tasks we may use the latent space to perform.
Conclusions & Future Directions
My research has moved in different directions since I ran these experiments, but I believe that advances in disentanglement learning will have useful applications in single cell genomics. Recent work has attempted to mitigate the negative effects of the trade-off I outlined above through different training methods1. Likewise, new research has suggested additional quantitative metrics that allow for more robust quantification of disentanglement. I look forward to seeing how this research progresses, and I would be happy to discuss future directions with any interested researchers!
Footnotes

Jacob C. Kimmel
Co-founder & President @ NewLimit. Interested in aging, genomics, imaging, & machine learning.